Major Contributions
IPOD:
a technology for global in vivo mapping of protein-DNA
interactions. We
developed a technology called In vivo protein occupancy
display (IPOD) that enables comprehensive monitoring of bacterial
transcriptional regulatory interactions. IPOD enables monitoring of
thousands of dynamic protein-DNA interaction sites across the genome,
revealing co-regulated regulons responsive to environmental and
genetic perturbations. By applying IPOD to E. coli, we
have
found that the E. coli chromosome contains hundreds of
kilo-base scale regions, bound by nucleoid proteins, that, through
their transcriptional silencing effect, appear to function as
prokaryotic analogs of eukaryotic heterochromatin.
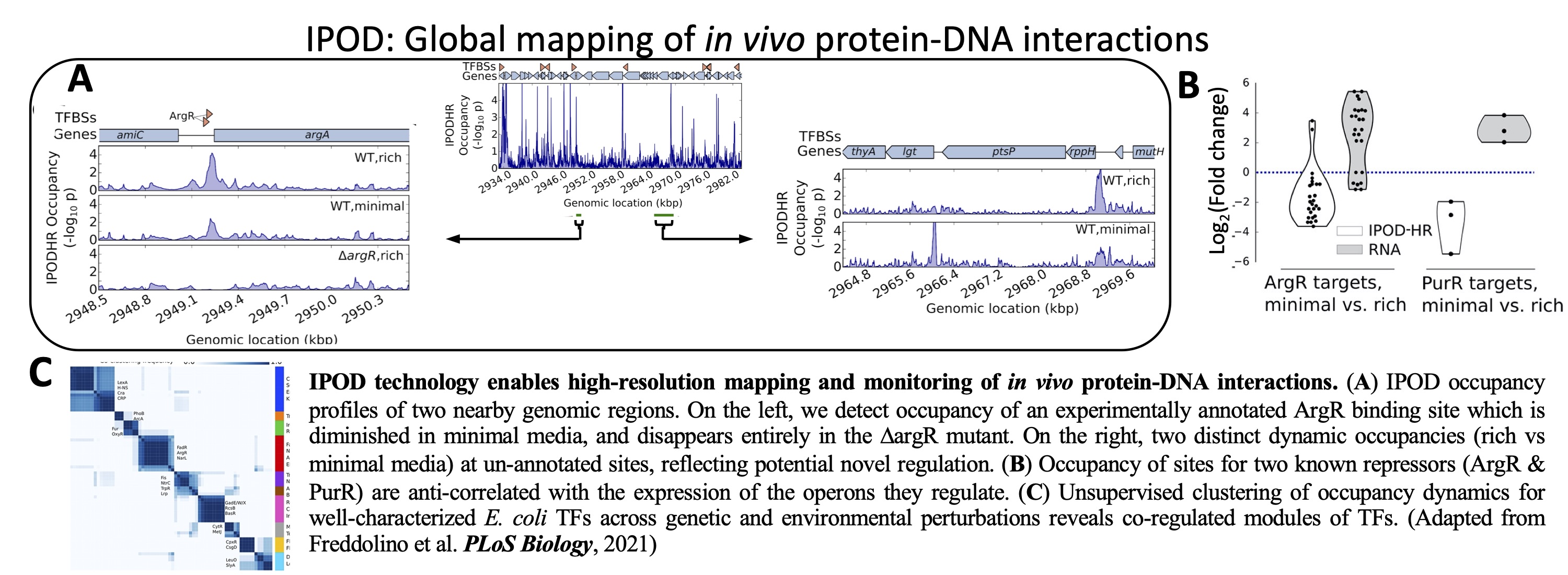
Dynamic landscape of protein occupancy across the Escherichia
coli chromosome.
PLoS
Biology
19(6):e3001306 2021 (bioRxiv Jan. 2020)
Freddolino, P., Amemiya, H.M., Goss, T.J., Tavazoie, S.
Global protein occupancy landscape of a bacterial genome
Molecular
Cell. 2009 Jul
31;35(2):247-53. PDF
Vora T, Hottes AK, Tavazoie S
In
vivo
mRNA display technology enables high-throughput proteomic
analysis by DNA-sequencing. We
developed
a technology called In vivo mRNA display that enables
high-throughput proteomics using next-generation sequencing as a
readout. This
is
achieved by coupling in vivo expressed proteins to their
encoding mRNAs using the high affinity interaction between a phage
coat protein and its cognate RNA stem-loop. This technology
bypasses the labor, cost, sensitivity, and throughput limitations of
mass-spectrometry to advance a variety of proteomics applications.
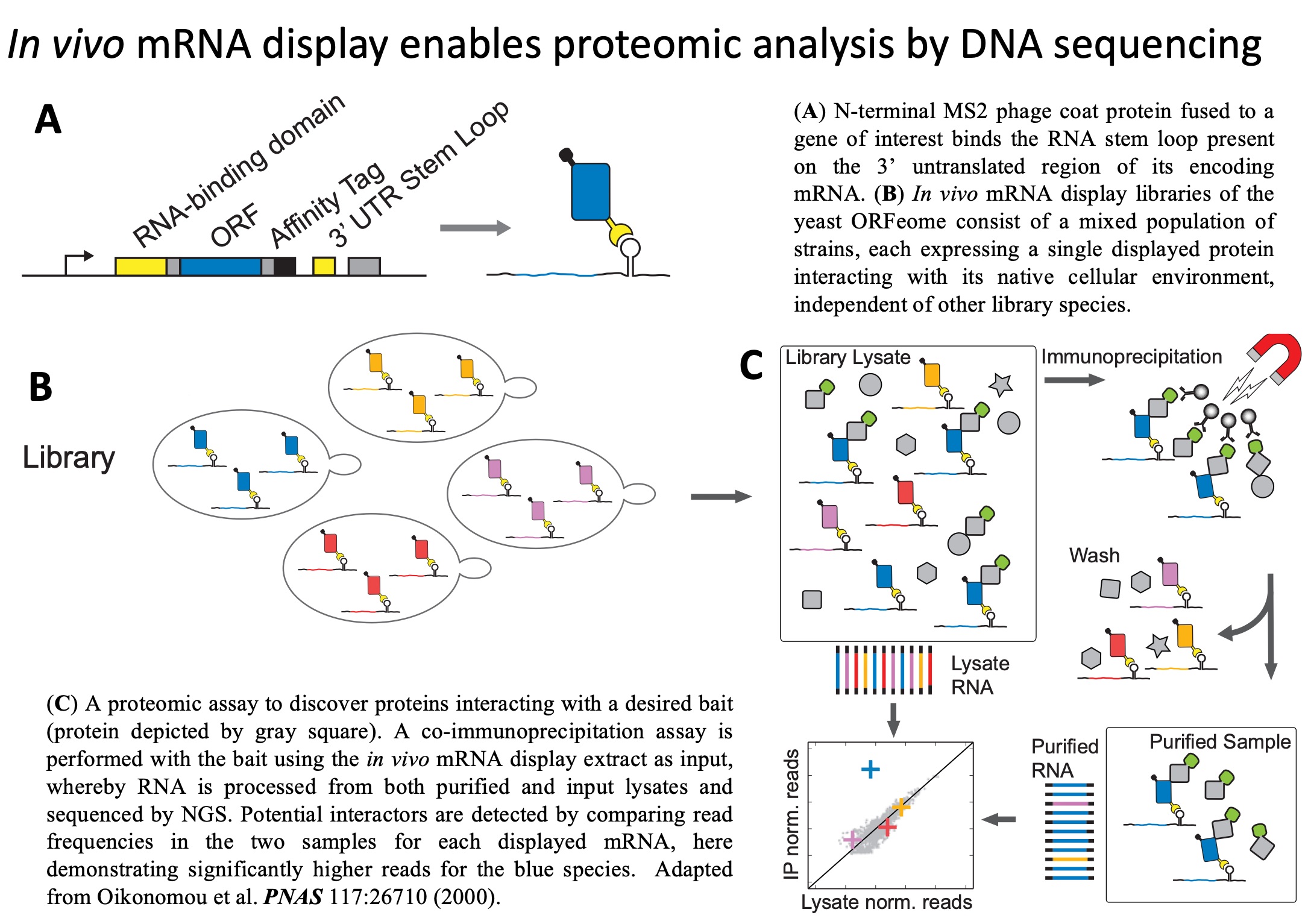
In vivo mRNA display enables large-scale proteomics by next
generation sequencing.
PNAS
117(43):26710
2020
Oikonomou, P., Salatino, R., Tavazoie, S.
PETRI-seq:
A technology for high-throughput bacterial single-cell RNA
sequencing. We developed PETRI-seq, the
first high-throughput prokaryotic single-cell RNA-sequencing
technology. PETRI-seq enables single-cell RNA sequencing of tens of
thousands of bacteria with capture-rates that permit identification
of rare cell-states.
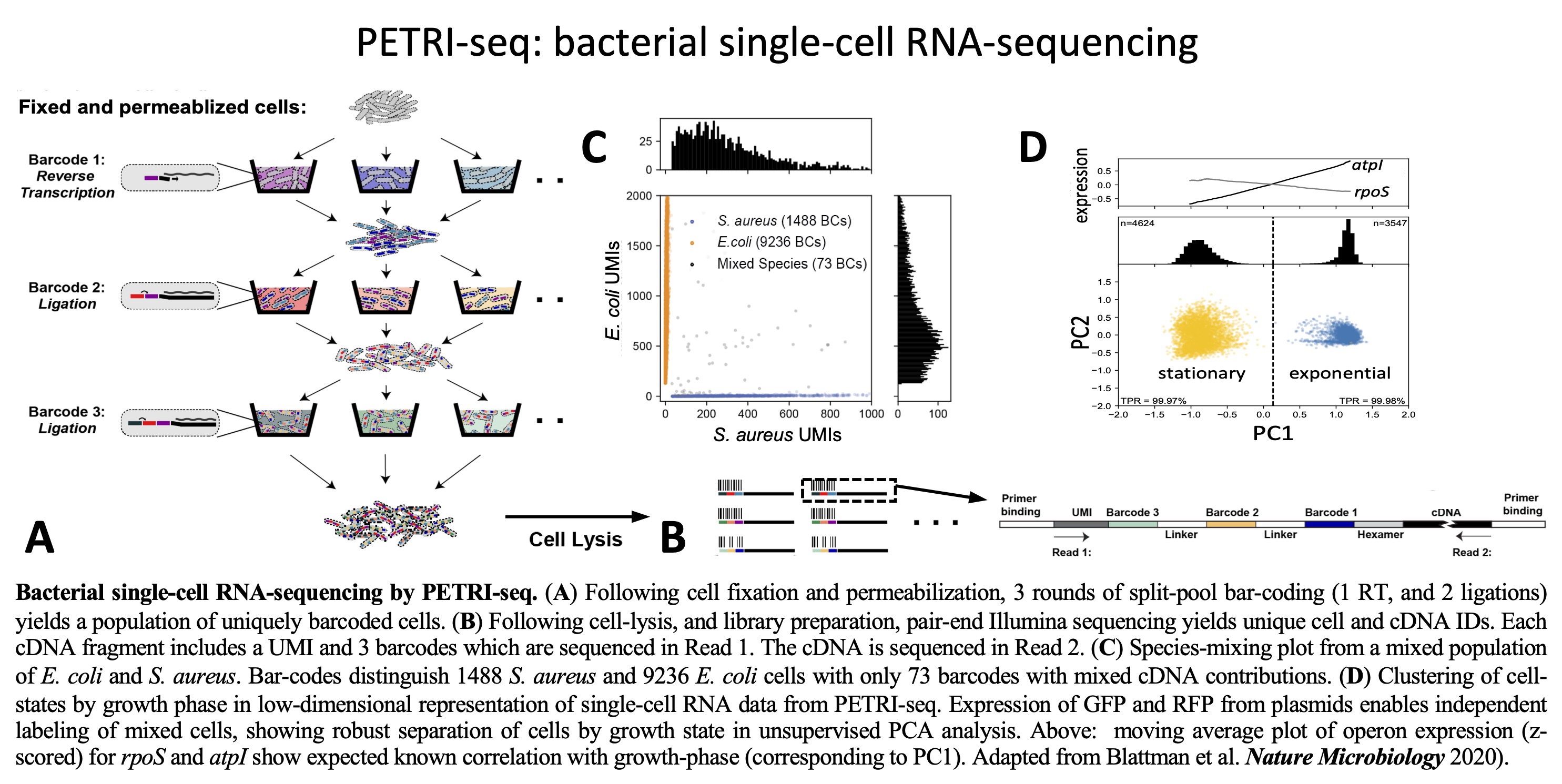
Prokaryotic Single-Cell RNA Sequencing by in Situ Combinatorial
Indexing.
Nature Microbiology May 25, 2020 (bioRxiv Dec. 6, 2019)
Blattman, S.B., Jiang, W., Oikonomou, P., Tavazoie, S.
CRISPR-interference
technology for rapid comprehensive genetic interrogation of
bacterial phenotypes. We developed a technology called CALM
that enables rapid generation of near-comprehensive
CRISPR-interference libraries in bacteria. The massive scale of CALM
libraries can produce a broad range of inhibition, providing high
sensitivity for mapping the genetic basis of bacterial phenotypes,
including the complex genetics of antibiotic sensitivity as
demonstrated here.
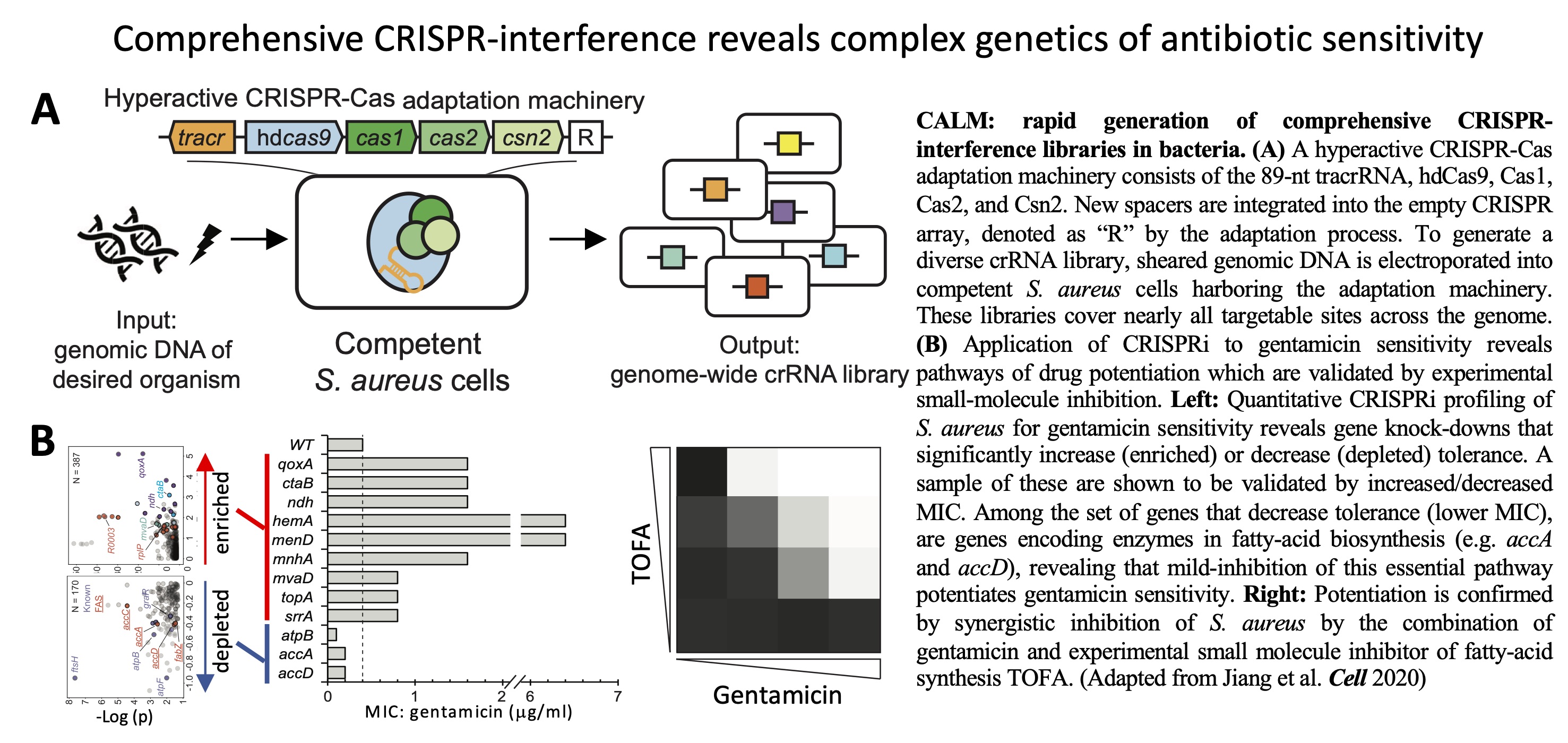
Comprehensive genome-wide perturbations via CRISPR adaptation
reveal complex genetics of antibiotic sensitivity.
Cell
180(5):1002 2020
Jiang, W, Oikonomou, P., Tavazoie, S.
Mutations to translation-related processes lead to extreme
antibiotic persistence. We
utilized laboratory evolution for antibiotic persistence in E.
coli to discover mutations that increase persistence frequency
to multiple antibiotics. This study revealed that mutations
targeting the process of translation result in population
heterogeneity and consequent >100-fold increase in persistence
rate.
Extreme Antibiotic Persistence via Heterogeneity-Generating
Mutations Targeting Translation.
mSystems
5(1), pii: e00847-19 Januray 21, 2020
Khare, A., Tavazoie, S.
Discovery
of
stochastic tuning: a mechanism for adaptive reprogramming of
gene expression by trial & error. Using a combination of theoretical and
experimental studies, we discovered a new principle of cellular
adaptation we call stochastic tuning. Stochastic tuning is a
versatile alternative to conventional gene regulation, enabling
cells to adapt to unfamiliar environments through active
reinforcement of random gene expression changes that improve
cellular health. Stochastic tuning may be a widespread phenomenon in
eukaryotes, enabling cells to optimize their gene expression states
beyond the precision possible with conventional regulatory
regulators such as transcription factors and RNA binding proteins.
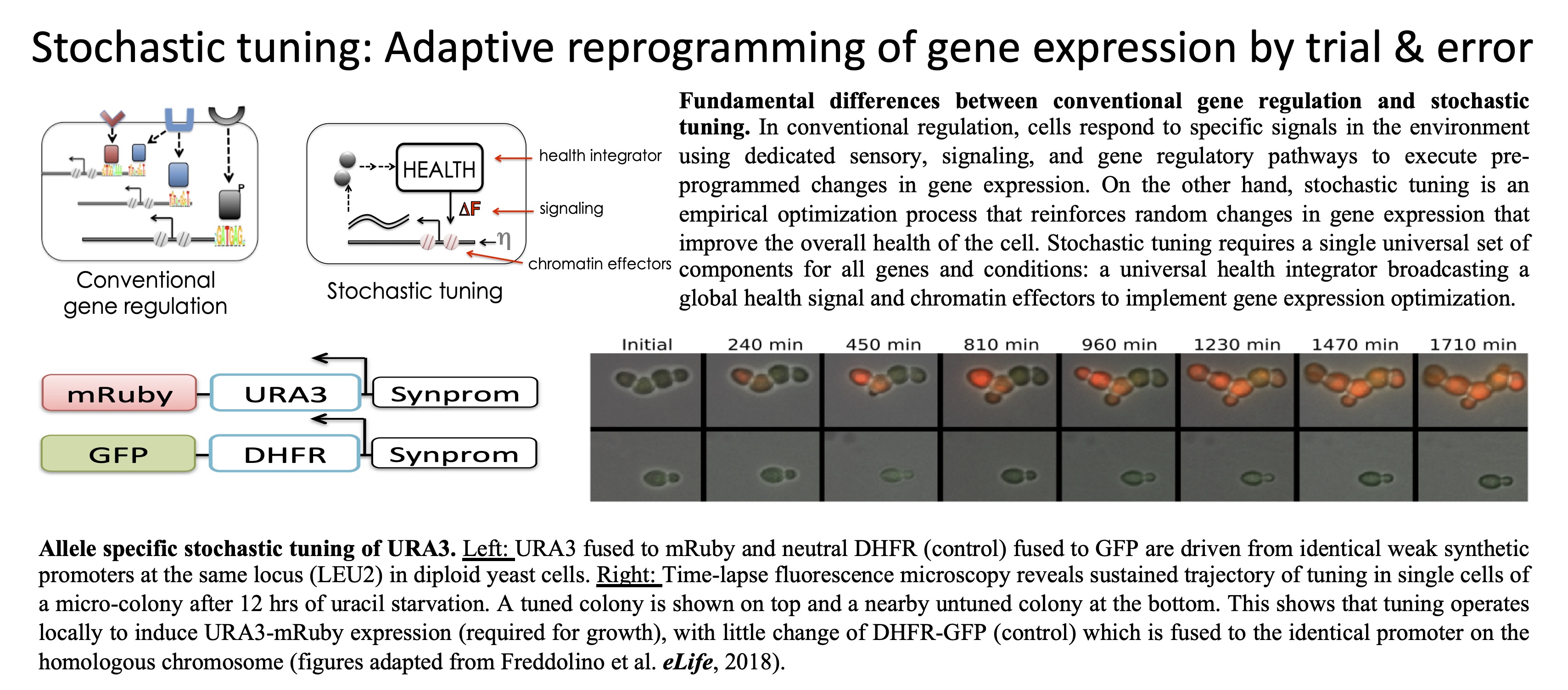
Stochastic tuning of gene expression enables cellular adaptation
in the absence of pre-existing regulatory circuitry.
eLife
2018;7:e31867 DOI: 10.7554/eLife.31867 (bioRxiv,
May 2017)
Freddolino, P., Yang, J., Momen-Roknabadi, A., Tavazoie, S.
A
systems biology framework for discovering the genetic
determinants of antagonism in multi-species bacterial
communities.
We used a set of unbiased genome-scale approaches to systematically
define the factors that contribute to antagonism in a two-species
model system of P. aeruginosa and E. coli. We found
that iron sequestration led to exploitative competition, while
phenazine exposure led to interference competition. Laboratory
evolution experiments revealed adaptive strategies to bypass these
strategies. This study demonstrates the power of agnostic systems
biology approaches in revealing the molecular basis of interference
and competition in multi-species communities.
Multifactorial competition and resistance in a two-species
bacterial system.
PLoS
Genetics
2015 11(12):e1005715
Khare,
A., Tavazoie, S.
Discovery
that
the RNA-binding protein HNRNPA2B1 is a nuclear reader of the
m(6)A modifications on mRNAs. In this collaborative work we find that the
RNA-binding protein HNRNPA2B1 binds m(6)A modified RNAs in vivo
within the nucleus and leads to similar alternative splicing
effects as perturbations to the m(6)A writer METTL3.
HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing
events.
Cell
2015 162(6):1299-308
Alarcon, C.R., Goodarzi, H., Lee, H, Liu, X., Tavazoie, S.,
Tavazoie, S.F.
Discovery
of
a structural RNA regulatory element controlling mRNA stability
and breast cancer metastasis.
We utilized a set of computational and experimental studies
to probe global transcriptomes and discover a structural RNA element
that regulates mRNA stability and breast cancer metastasis through
binding of the RNA binding protein TARBP2.
Metastasis-suppressor transcript destabilization through TARBP2
binding of mRNA hairpins.
Nature.
2014 513, 255-260
Goodarzi, H., Zhang, S., Buss, C.G., Fish, L., Tavazoie, S., &
Tavazoie, S.F.
High-throughput
profiling
of conserved human 3’UTR sequences for post-transcriptional
activity. We
generated
a fluorescent reporter library of conserved human 3’UTR sequences
and used FAC-sorting and next-generation sequencing to
comprehensively profile their post-transcriptional contributions.
This led to the discovery of a large set of linear and structural
RNA elements that post-transcriptionally modulate the fates of human
transcripts.
Systematic Identification of Regulatory Elements in Conserved 3′
UTRs of Human Transcripts.
Cell
Reports. 2014 Mar
20.
Oikonomou, P., Goodarzi, H. & Tavazoie, S.
Synaptic
state
matching: a new mechanism of synaptic potentiation that enables
self-supervised predictive learning in neural networks.
In this theoretical work, we introduced the principle of ‘synaptic
state matching’(SSM) which is a local, biologically plausible
mechanism for synaptic potentiation and homeostasis in neural
networks. We show that SSM, implemented in artificial neural
networks, enables generation of stable predictive internal
representations, leading to pattern completion, sequence-learning
and unsupervised feature detection. SSM is a biologically plausible
mechanism for the ‘predictive coding hypothesis’ and prescribes the
low-level hardware required for self-supervised learning in natural
and artificial intelligence systems.
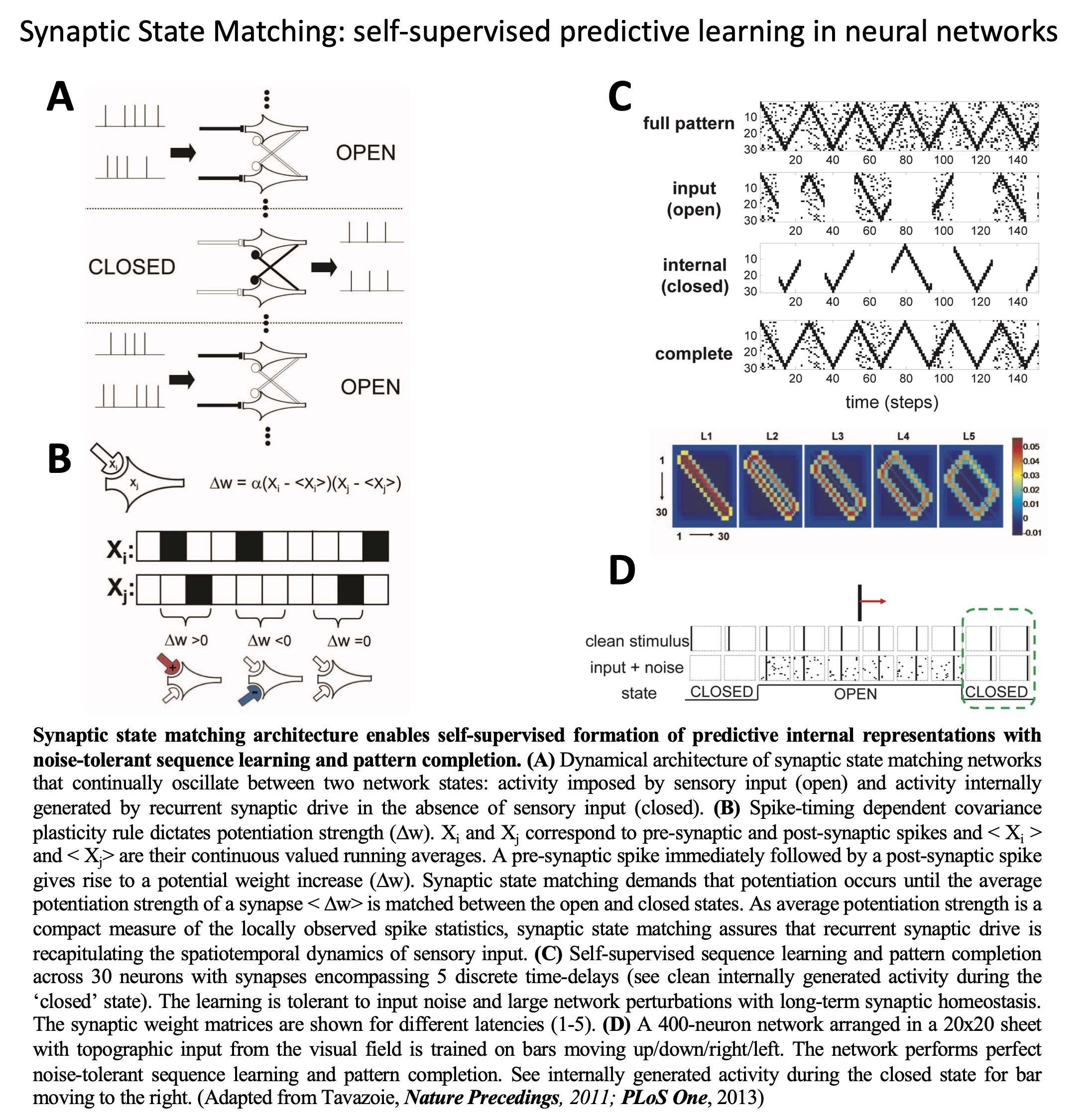
Synaptic state matching: a dynamical architecture for predictive
internal representation and feature detection.
PLoS
One. 2013 Aug
26;8(8):e72865. doi: 10.1371/journal.pone.0072865.
Tavazoie, S.
Synaptic state matching: a dynamical architecture for predictive
internal representation and feature perception.
Nature
precedings
(Aug 2011)
Tavazoie, S.
Rapid
adaptation
of bacteria to extreme environments through loss-of-function
mutations.
In this work, we compiled a set of diverse transposon-profiling
experiments, previously published by our group, to show that
loss-of-function mutations are a substantial contributor to
adaptation of bacteria to extreme environments. Using these
observations, we argue that often fitness bottlenecks are caused by
regulatory and metabolic constraints that can be easily bypassed by
loss-of-function of individual regulatory genes. This work has
implications for diverse areas of investigation including emergence
of antibiotic resistance, understanding limitations of unculturable
bacteria, and engineering of synthetic microbes for extreme
environments.
Bacterial adaptation through loss of function.
PLoS
Genetics.
2013;9(7):e1003617. doi: 10.1371/journal.pgen.1003617.
Hottes, A.K., Freddolino, P.L., Khare, A., Donnell, Z.N., Liu, J.C.,
& Tavazoie, S.
Large
increases
in antibiotic persistence through dozens of easily acquired
mutations.
We used transposon mutagenesis and profiling to show that there
exist dozens of genes in the E. coli genome that when
disrupted cause substantially increased antibiotic persistence. The
functions of these loci are informative of common mechanisms of
persistence. The large number of loci and the ease with which
mutations in them can cause loss of function suggest that rapid
evolution of increased persistence-rate may be a common process of
critical significance in the setting of therapeutic tolerance to
antibiotics.
Large mutational target size for rapid emergence of bacterial
persistence.
PNAS.
2012 Jul 16. [Epub ahead of print] PDF
Girgis, H.S., Harris, K., & Tavazoie, S.
A
single amino-acid mutation causes a dramatic global shift in the
fitness landscape of E. coli.
Through extensive phenotypic analyses, we have characterized the
systems-level consequences of a missense mutation in the global
transcriptional terminator Rho of E. coli. We find that a
single amino acid change in Rho results in a massive change in the
fitness landscape of the cell, with widely discrepant fitness
consequences of identical single locus mutations in rho* versus rho
WT backgrounds. Our observations reveal the extent to which a single
regulatory mutation can transform the entire fitness landscape of
the cell, causing a massive change in the interpretation of
individual mutations and altering the evolutionary trajectories
which may be accessible to a bacterial population.
Fitness landscape transformation through a single amino acid
change in the Rho terminator
PLoS
Genetics, Vol. 8,
No. 5. (2012), e1002744
Freddolino, P., Goodarzi, H., & Tavazoie, S.
TEISER:
a
structural RNA motif finder that enables transcriptome-wide
discovery of post-transcriptional regulatory elements.
In this work, we demonstrate the discovery of structural RNA
regulatory elements that underlie post-transcriptional regulation of
gene expression in mammalian genomes. To accomplish this, we
developed a new algorithm for de novo discovery of
structural RNA regulatory elements (TEISER) that utilizes
context-free grammar representations that capture both sequence and
structure. We take one of the top predictions of an mRNA stability
element and using biochemistry, mass spectrometry and in vivo
binding studies, show that it is bound by HNRPA2B1, a human RNA
binding protein that binds this element and stabilizes a large
number of its target genes. Our approach has revealed the putative
involvement of a large family of structural RNA elements that
control various fates of mRNA, including stability, splicing,
localization, and translation.
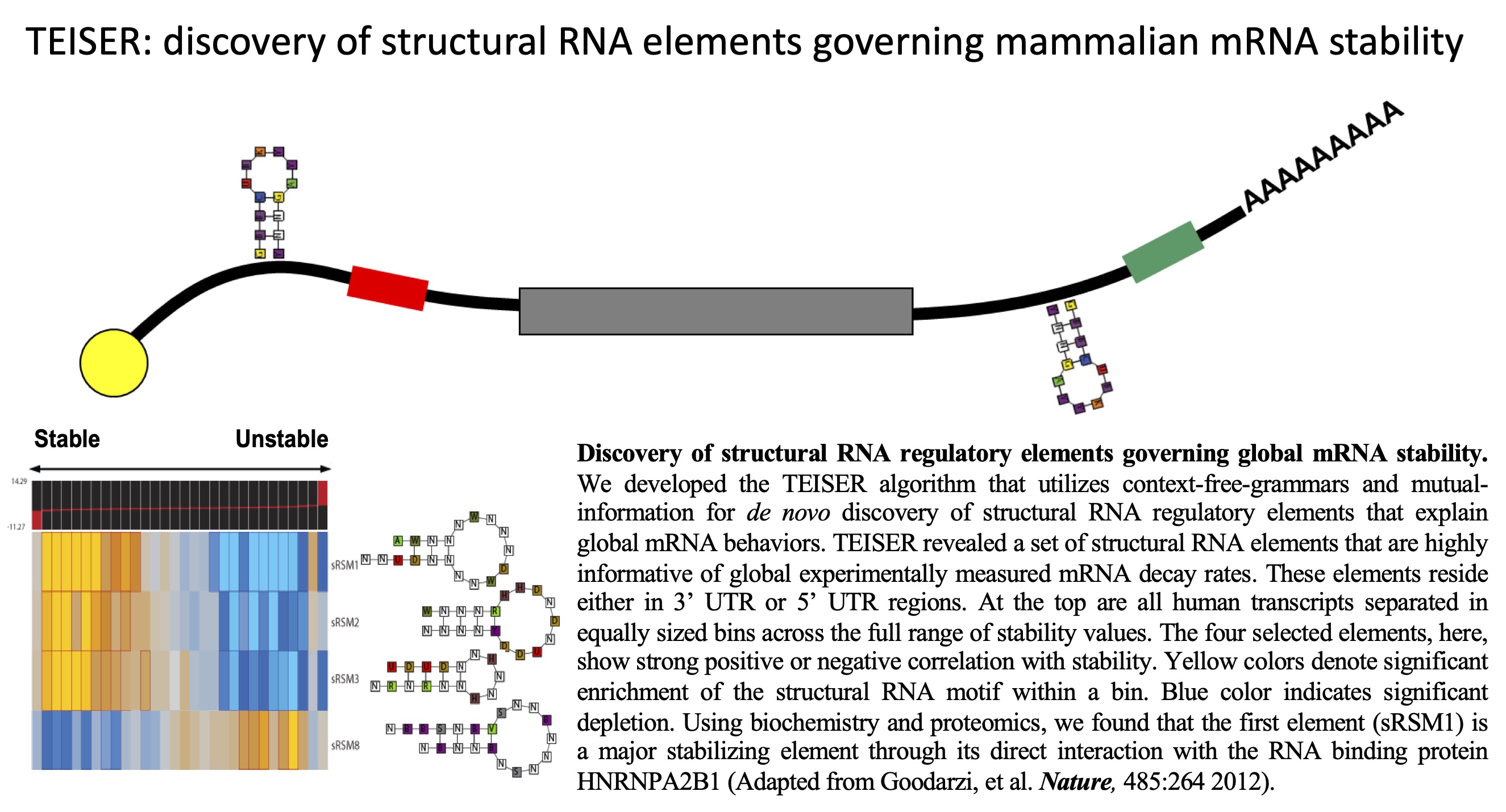
Systematic discovery of structural elements governing stability
of mammalian messenger RNAs
Nature
485, 264-268 (2012) PDF
Goodarzi, H., Najafabadi, H.S., Oikonomou, P., Greco, T.M., Fish,
L., Salavati, R., Cristea, I.M., & Tavazoie, S.
Fitness
profiling
and experimental evolution reveal regulatory and metabolic
adaptation of E. coli to a severe stress.
In this work, we utilized global transposon profiling to understand
the genetic basis of ethanol tolerance in E. coli. Among
multiple adaptive pathways of osmoregulation and cell-wall
biogenesis, we also discovered that loss-of-function mutations in
specific regulators have the potential of reversing the flux through
the TCA, leading to ethanol detoxification. By performing laboratory
evolution experiments and phenotypic and metabolic analyses, we show
that this hypothetical solution is actually utilized by E. coli
in practice, leading to ethanol degradation by the TCA. This work
demonstrates the flexibility of regulatory and metabolic networks in
rewiring cell metabolism to enable rapid adaptation to extreme
environments.
Regulatory and metabolic rewiring during laboratory evolution of
ethanol tolerance in E. coli
Mol
Syst Biol. 2010
Jun 8; 6:378. PDF
Goodarzi H, Bennett BD, Amini S, Reaves ML, Hottes AK, Rabinowitz
JD, Tavazoie S
Systematic
discovery
of regulatory and pathway perturbations across large cancer
datasets.
In this work, we describe the development and application of
algorithms that reveal the transcriptional and post-transcriptional
regulatory networks that are dysregulated across large cohorts of
human cancer transcriptomes. Our information-theoretic analyses,
across a diverse set of human cancers, revealed the majority of
previously known cancer pathways along with many more novel
predictions encompassing DNA and RNA regulatory elements and
pathways. These analyses and the accompanying experimental
validations demonstrate that agnostic systems biology approaches can
be used to systematically discover regulatory networks and pathways
that underlie oncogenesis and oncogenic progression.

Revealing global regulatory perturbations across human cancers
Molecular
Cell. 2009 Dec
11; 36:900-911. PDF
Goodarzi, H, Elemento, O, Tavazoie S
Systematic
genetic
dissection of bacterial phenotypes within biofilms. In
these studies, we devised a global transposon-based approach to
determine the genetic basis of biofilm formation (E. coli)
and biofilm-dependent antibiotic tolerance (p. aeruginosa).
These studies revealed biophysical and biochemical insights into the
unique nature of bacterial life within biofilms and the power of a
global systematic approach for rapidly determining the genetic basis
of complex phenotypes.
Genetic
dissection
of an exogenously induced biofilm in laboratory and clinical
isolates of E. coli.
PLoS
Pathogens 2009 May;5(5):e1000432. Epub 2009 May 15. PDF
Amini S, Goodarzi H, Tavazoie S
Fitness
Landscape
of Antibiotic Tolerance in Pseudomonas aeruginosa Biofilms
PLoS
Pathogens 7(10):e1002298 (2011). PDF
Amini S., Hottes, A.K., Tavazoie, S.
ADAM:
a
technology for rapid discovery of adaptive mutations
contributing to bacterial fitness.
In this work, we describe the development of a technology (ADAM) for
systematically identifying the subset of adaptive mutations that
contribute to changes in fitness in laboratory evolved bacteria.
This technology alleviates the labor-intensive task of separating
driver and passenger mutations in a variety of natural and
biomedically driven evolutionary trajectories.
Global discovery of adaptive mutations
Nature
Methods. 2009
Aug; 6(8):581-3. PDF
Goodarzi H, Hottes AK, Tavazoie S
Discovery
of
predictive behavior in E. coli.
This is the first demonstration of predictive behavior by
microorganisms. Using a combination of theoretical, simulation, and
experimental work, we show that the biochemical networks of the
bacterium E. coli are able to carry out predictive behavior
akin to animal nervous systems. This work was motivated by the
observation that microbial habitats are highly structured in space
and time and therefore the perception of change in one parameter
(e.g. increase in temperature) can be highly predictive of an
impending change in another parameter (e.g. decrease in oxygen).
Utilizing an in silico ecology,
we showed that such dynamically structured environments give rise to
the evolution of complex biochemical networks that predict the
short-term trajectory of their environments. We provided
experimental evidence for such anticipatory behavior in responses of
E. coli to changes in
temperature and oxygen that mirror transitions between the outside
environment and the mammalian gastrointestinal tract. Using
laboratory evolution, we further demonstrated that circuits
controlling predictive behavior can be rewired by exposing E.
coli to dynamic environments with the opposite correlation to
that of the native habitat. Predictive behavior in microbes has
since been demonstrated in other microbes, including yeast,
suggesting that it is a widespread phenomenon. These studies have
fundamental implications for our understanding of microbial
behaviors, and have reshaped even the most basic interpretation of
laboratory experiments.

Predictive behavior within microbial genetic networks
Science
(2008) 320:1313-1317, Epub 2008 May 8. PDF
Tagkopoulos I, Liu Y, Tavazoie S
Beyond homeostasis: a predictive-dynamic framework for
understanding cellular behavior.
Annu
Rev Cell Biol
2012. Vol. 28: 363-384
Freddolino, P.L., & Tavazoie, S.
FIRE:
an
information theoretic algorithm for sensitive discovery of DNA
and RNA regulatory elements across gene expression datasets. In this work, we
introduce a novel information-theoretic approach for discovering DNA
and RNA regulatory elements that underlie global changes in gene
expression. Our approach performs a course-grained comprehensive
search for DNA and RNA sequence motifs that have high mutual
information with large-scale observations of gene expression. In
doing so, we are able to discover regulatory elements with
exceptionally high sensitivity and near zero false discovery rates
across organisms ranging from bacteria to human. Our algorithm is
implemented in the FIRE (Finding Informative Regulatory Elements)
software, available for local or server-based use (https://tavazoielab.c2b2.columbia.edu/FIRE).
FIRE
has been used by hundreds of independent studies to reveal
transcriptional and post-transcriptional regulatory programs
underlying diverse physiological, developmental, and pathological
processes.
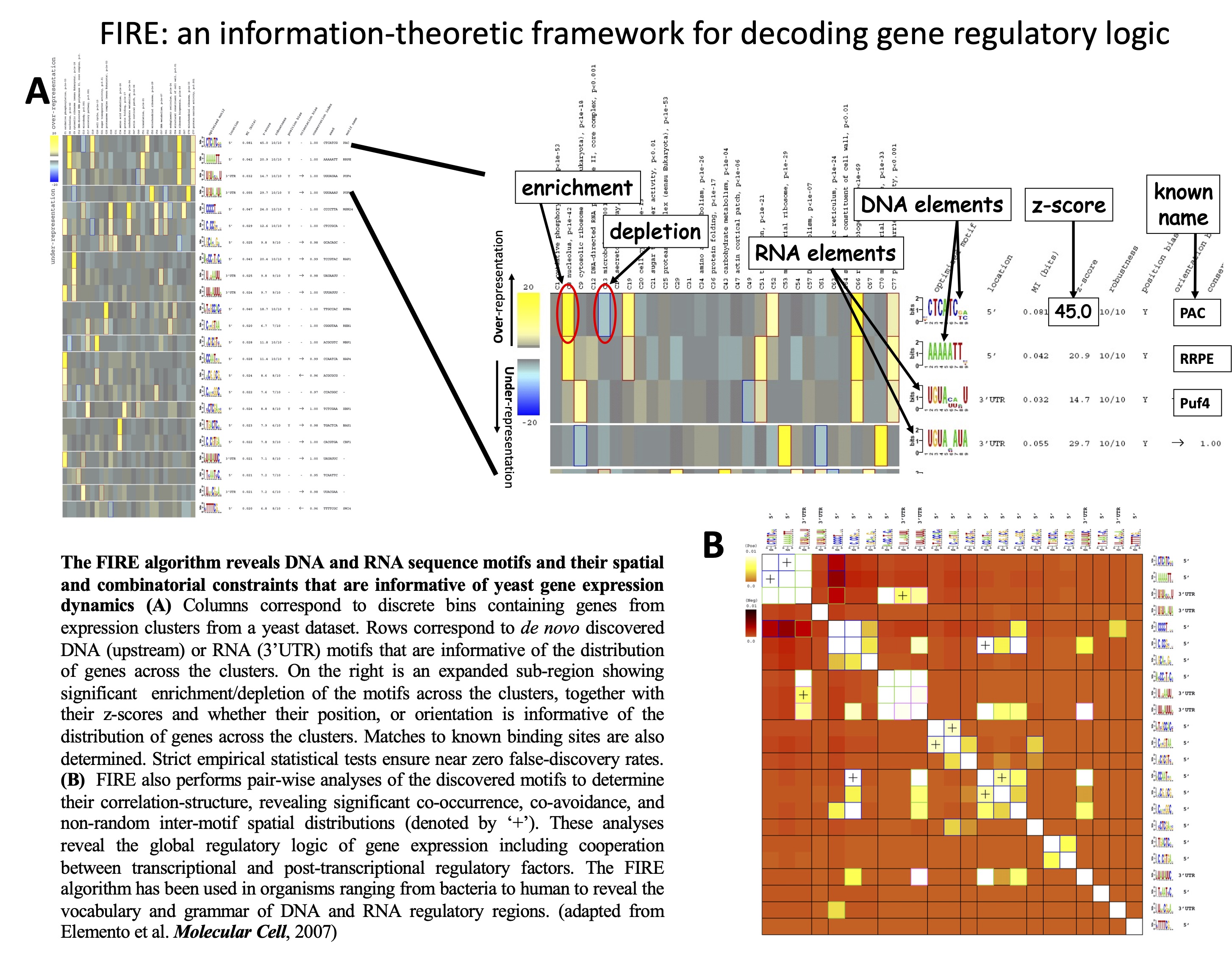
A universal framework for regulatory element discovery across all
genomes and data-types
Molecular
Cell (2007)
28(2):337-50. PDF
Elemento
O, Slonim N, Tavazoie, S
Revealing global regulatory perturbations across human cancers
Molecular
Cell. 2009 Dec
11; 36:900-911. PDF
Goodarzi, H, Elemento, O, Tavazoie S
Molecular topography of an entire nervous system.
Cell
Jul 2:S0092-8674(21)00758-3 2021
Taylor, S.R. et al.
Systematic
interrogation
of gene function and epistasis in bacterial chemotaxis using
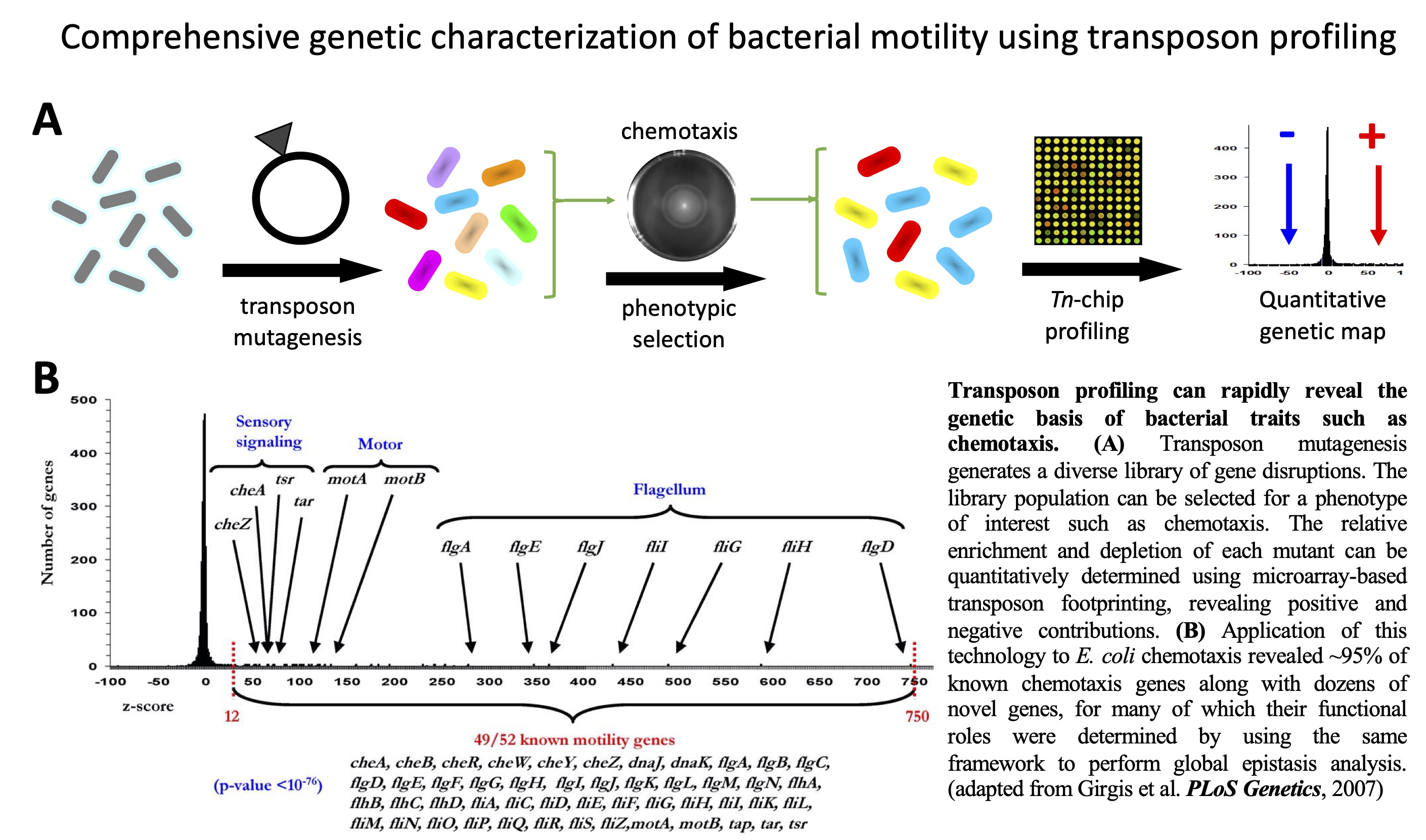
transposon profiling. Here,
we introduced an efficient framework for rapidly discovering the
genetic basis of bacterial traits through the power of deep
transposon mutagenesis, library selection, and parallel readout of
transposon mutant spectra through microarray hybridization. In order
to characterize the sensitivity of this technology, we applied it to
one of the best understood bacterial behaviors-flagella mediated
chemotaxis. Our strategy allowed a single graduate student to
systematically survey the contribution of every gene in the E. coli genome to chemotaxis within the time-scale of a few weeks.
This led to the identification of 95% of the roughly 50 known
flagella and chemotaxis genes. Remarkably, we also discovered three
dozen additional genes that clearly impact motility, many of which
were genes of previously unknown function. By utilizing deep
transposon libraries in the background of individual mutants, we
were able to perform global epistasis analysis and place many of
these novel genes within the context of known biochemical and
regulatory pathways, including LPS signaling and cyclic di-GMP
metabolism. The success of our systematic perturbation approach in
revealing the genetic basis of a complex phenotype laid the
groundwork for the development of many advances in the field such as
CRISPR-based perturbation libraries that came years later.
A comprehensive genetic characterization of bacterial motility
PLoS
Genetics (2007) 3
(9): e154. PDF
Girgis H, Liu Y, Ryu W, Tavazoie S
Computational
discovery
of genes and gene modules underlying microbial traits.
In this work, we introduce a computational framework that reveals
the modular genetic architecture of bacterial traits by correlating
the cross-species distribution of genes and phenotypes.
At the core of our approach is the simple assumption that
proteins required for the expression of a phenotype are
statistically enriched in the organisms that manifest the phenotype.
Since our approach does not require any prior
knowledge of genes involved, it is ideal for determining the genetic
basis of bacterial traits that are entirely uncharacterized.
We used information-theory to measure correlations between
phylogenetic profiles (a vector of ones [zeros] for presence
[absence], of a gene across the genomes), and phenotypic profiles (a
vector of ones [zeros] for the presence [absence], of a trait across
the genomes). This yielded a
highly sensitive framework that allowed successful genetic
characterization of a variety of traits such as
swimming motility, sporulation, and cellular morphology.
In addition to
identifying the components of these modules, we showed that
preferential co-inheritance of subsets of genes within them allows
us to organize them into functional sub-modules.
For example, in the case of flagellar-mediated chemotaxis,
our approach automatically revealed three independent sub-modules:
the flagellar apparatus, the chemotaxis sensory/signal
transduction network, and L/P rings which anchor the flagellum
into the outer membrane of gram-negative bacteria, but which are
absent from gram-positives. With
the massive increase in the rate of genome sequencing, approaches
like the one introduced here allows efficient and rapid
determination of genotype/phenotype relationships across diverse
bacterial species of basic and clinical importance.
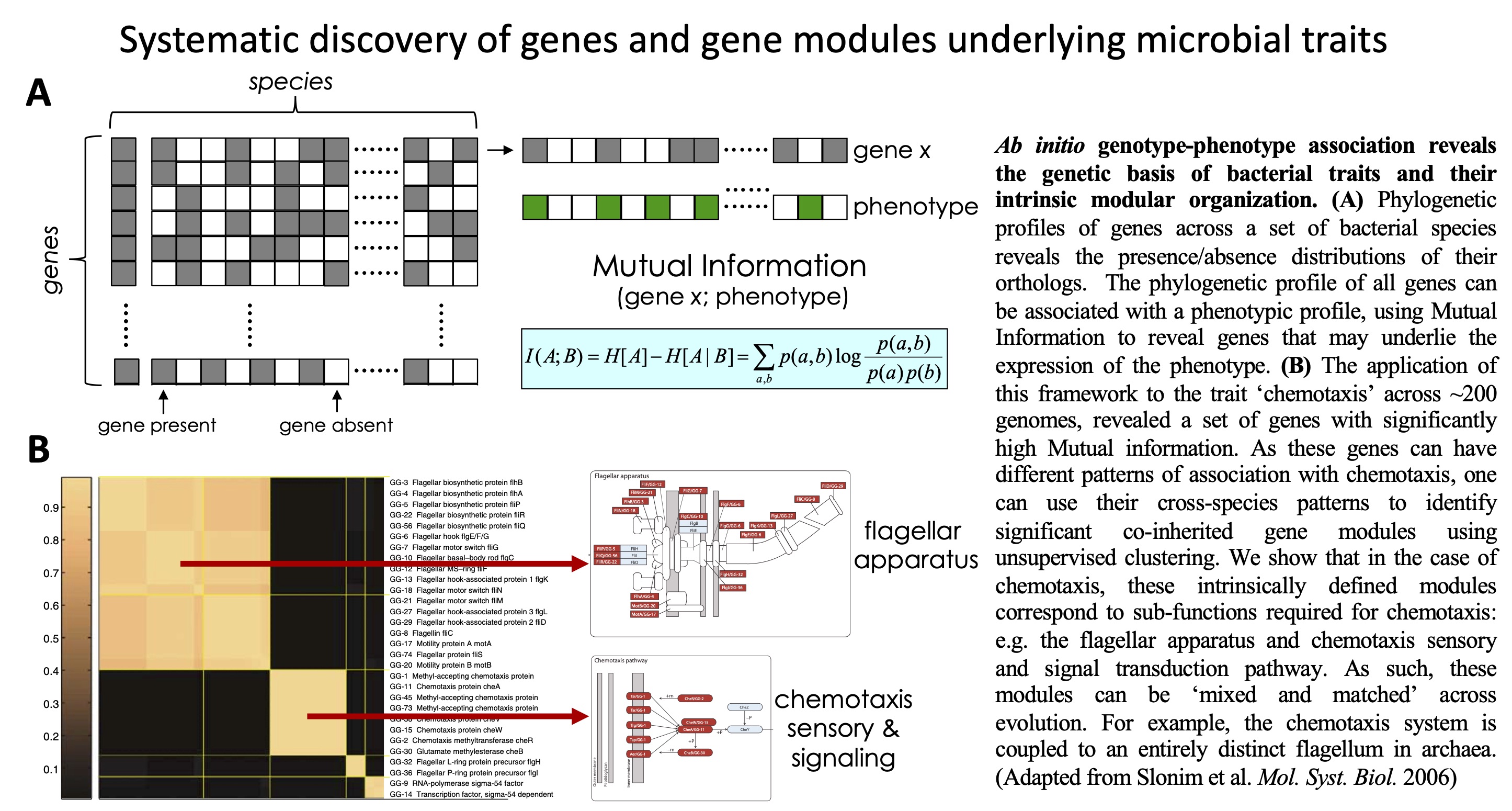
Ab initio genotype-phenotype
association reveals the intrinsic modularity of genetic networks.
Molecular
Systems Biology
2006; 2:2006.0005. Epub 2006 Jan 31. PDF
(Slonim N, Elemento O, equal contribution) and Tavazoie S
A cross-genomic approach for systematic mapping of phenotypic
traits to genes.
Genome
Research 2004
Jan; 14(1):109-15. PDF
Jim K, Parmar K, Singh M, Tavazoie S
Network-level
conservation
enables genome-wide discovery of conserved DNA/RNA regulatory
elements without sequence alignment. A powerful approach for
discovering DNA regulatory elements is to look for their
conservation between closely related species.
Usually, such elements are identified through comparisons of
individual genomic loci using alignments of regulatory regions from
multiple species. In these
studies, we introduced a novel concept called ‘network-level
conservation’ that extends this one-gene at a time comparison to the
whole genome, by demanding that the network
of regulatory interactions across the entire genome be conserved.
This strategy gave us unprecedented sensitivity to detect
functional genomic elements, without the need for alignments, while
requiring only two genomes. Using
network-level conservation, we generated comprehensive catalogues of
regulatory elements for many model organisms including the very
first global inventory of conserved elements between human and
mouse. These features also
allowed us to compare regulatory element catalogues between distant
phylogenetic groups. Although
we found many of the binding sites to be highly conserved in these
comparisons, strikingly, the genes regulated by these elements are
entirely different, and do not overlap any more than would be
expected by chance. In this
way, our work revealed that there is extensive re-wiring of
transcriptional networks even between relatively closely related
species. Subsequently, we
found such re-wiring to also be a dominant feature in the evolution
of post-transcriptional regulatory elements, including targets of
microRNAs and RNA-binding proteins.
Whole-genome discovery of transcription factor binding sites by
network-level conservation.
Genome
Research 2004
Jan; 14(1):99-108. PDF
Pritsker M, Liu Y, Beer M, Tavazoie S
Fast and systematic genome-wide discovery of conserved regulatory
elements using a non-alignment based approach.
Genome
Biology (2005)
6(2):R18. Epub 2005 Jan 26. PDF
Elemento O, Tavazoie S
Revealing posttranscriptional regulatory elements through
network-level conservation.
PLoS
Computational Biology
2005 Dec; 1(7): e69 Epub 2005 Dec. 9. PDF
(Chan S, Elemento O, equal contribution) and Tavazoie S
Patterns
of
histone acetylation are predictive of gene expression dynamics
in yeast.
In eukaryotes, DNA is wrapped around histone proteins (within
chromatin), and enzymatic modifications of histone tails is known to
be important for many processes that operate on DNA—including
transcription and DNA replication. In
collaboration with Michael Grunstein’s laboratory, we systematically
explored the extent to which modifications (acetylation) of these
histone tails is associated with changes in gene expression.
We observed that the patterns of multiple such
modifications convey unique information about the expression state
of genes. Our
observations are consistent with one dimension of the “histone code
hypothesis”, namely that consistent modification patterns code for
specific processes that are orchestrated on chromatin in order to
affect processes such as transcription or DNA replication.
Mapping global histone acetylation patterns to gene expression.
Cell
2004 Jun 11; 117(6):721-33. PDF
Kurdistani SK, Tavazoie S, Grunstein M
The
first
demonstration of a machine-learning strategy to decode DNA
regulatory logic, enabling prediction of gene expression
dynamics from sequence alone.
In this work, we introduced a computational framework that allows
predictive modeling of gene expression dynamics through
machine-learning models that map sequence to gene expression. This
approach was the first to successfully predict global gene
expression patterns based solely on local DNA sequences. This
allowed us not only to predict the expression pattern of genes, but
also to assess the extent to which the information within local DNA
influenced gene expression patterns across microarray experiments.
We designed our approach to function in a largely unbiased fashion
with respect to previous biological knowledge, using only microarray
expression data, genomic sequence, and minimal constraints. This
systematic strategy revealed DNA regulatory elements that, in a
combinatorial fashion, operate to affect gene-expression. We also
discovered that the functionality of many of these elements requires
precise spatial and pair-wise configurational constraints within
regulatory regions. These findings addressed a major conundrum in
the field: what determines the functional context of
transcription-factor binding sites? Our ability to achieve robust
predictions for >70% of genes established that, at least in
yeast, most of the regulatory information resides in the ~800
base-pairs 5’ upstream of the genes. In this work, we went further
to show that the same framework can successfully reveal DNA
regulatory logic underlying temporal gene expression dynamics during
C. elegans development. This work established that a sufficiently
constrained machine-learning framework can reveal DNA cis-regulatory programs on a genomic scale.
As such, this work greatly influenced subsequent work in the
field, and laid the foundation for systematic elucidation of such
regulatory programs in mammalian genomes.
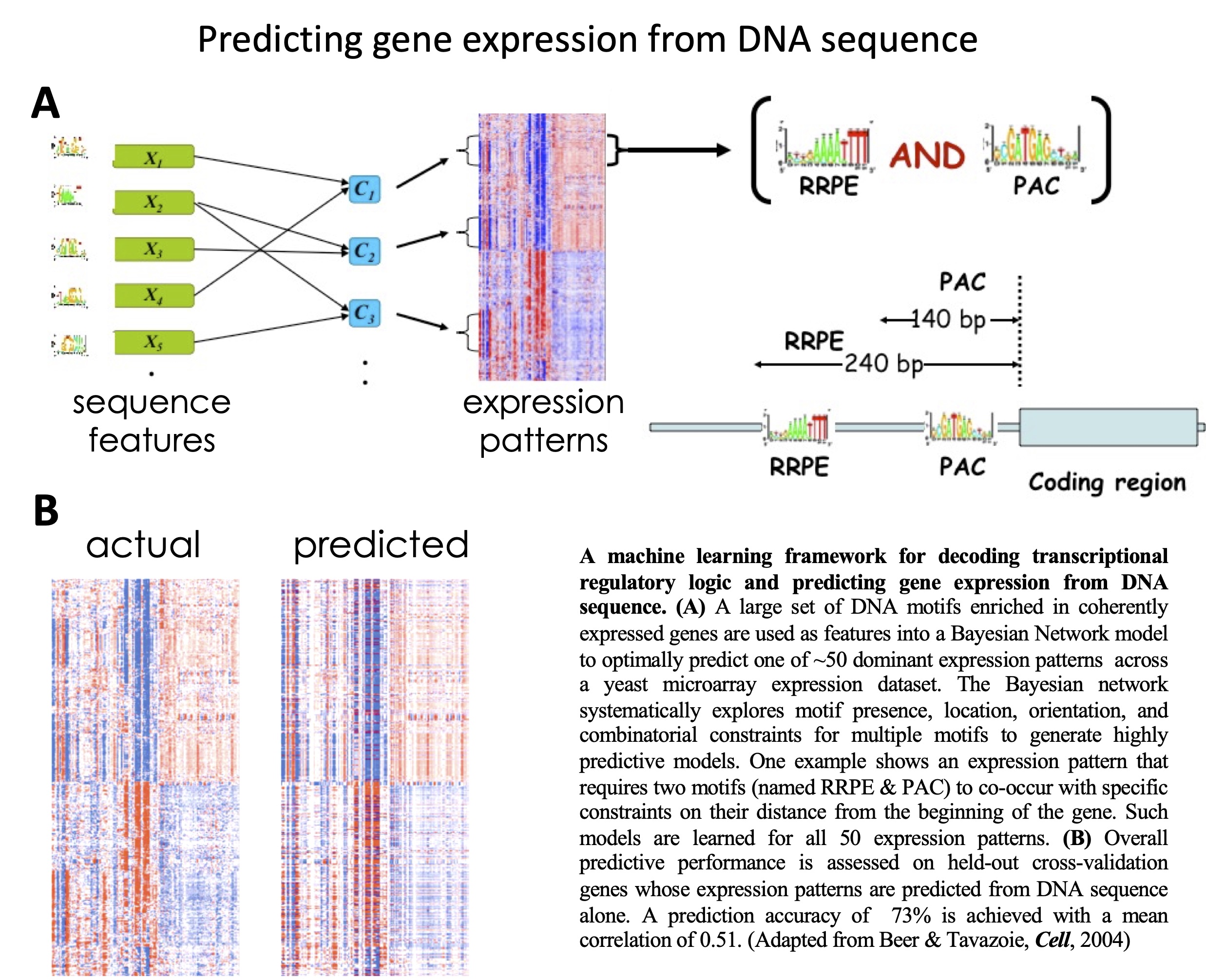
Predicting gene expression from sequence.
Cell
2004 Apr 16; 117(2):185-98. PDF
Beer MA, Tavazoie S
Systematic
discovery
of DNA regulatory elements and pathways underlying gene
expression dynamics across the yeast cell-cycle. This study presented one of
the first systematic analyses of microarray expression data.
Here, we implemented a novel machine-learning framework that
utilized microarray expression data and genomic sequence information
to identify co-expressed sets of genes and the DNA regulatory
elements that implement their co-regulation. This approach revealed
most of the known cell-cycle regulatory transcription factor binding
sites in yeast, along with novel predictions that have since been
experimentally validated by us and others.
The computational framework we introduced here was
subsequently adopted and extended broadly by the community, leading
to thousands of follow-up publications.
These analyses included: discovering co-expressed regulons by
clustering of expression data, discovery and characterization of
regulatory motifs in co-expressed genes, and statistical enrichment
of gene-sets for functional categories, a predecessor to gene-set
enrichment analysis. More broadly, we demonstrated that a largely
agnostic machine-learning framework can be used to systematically
reverse-engineer the regulatory network of a complex process,
recapitulating decades of work across dozens of labs.
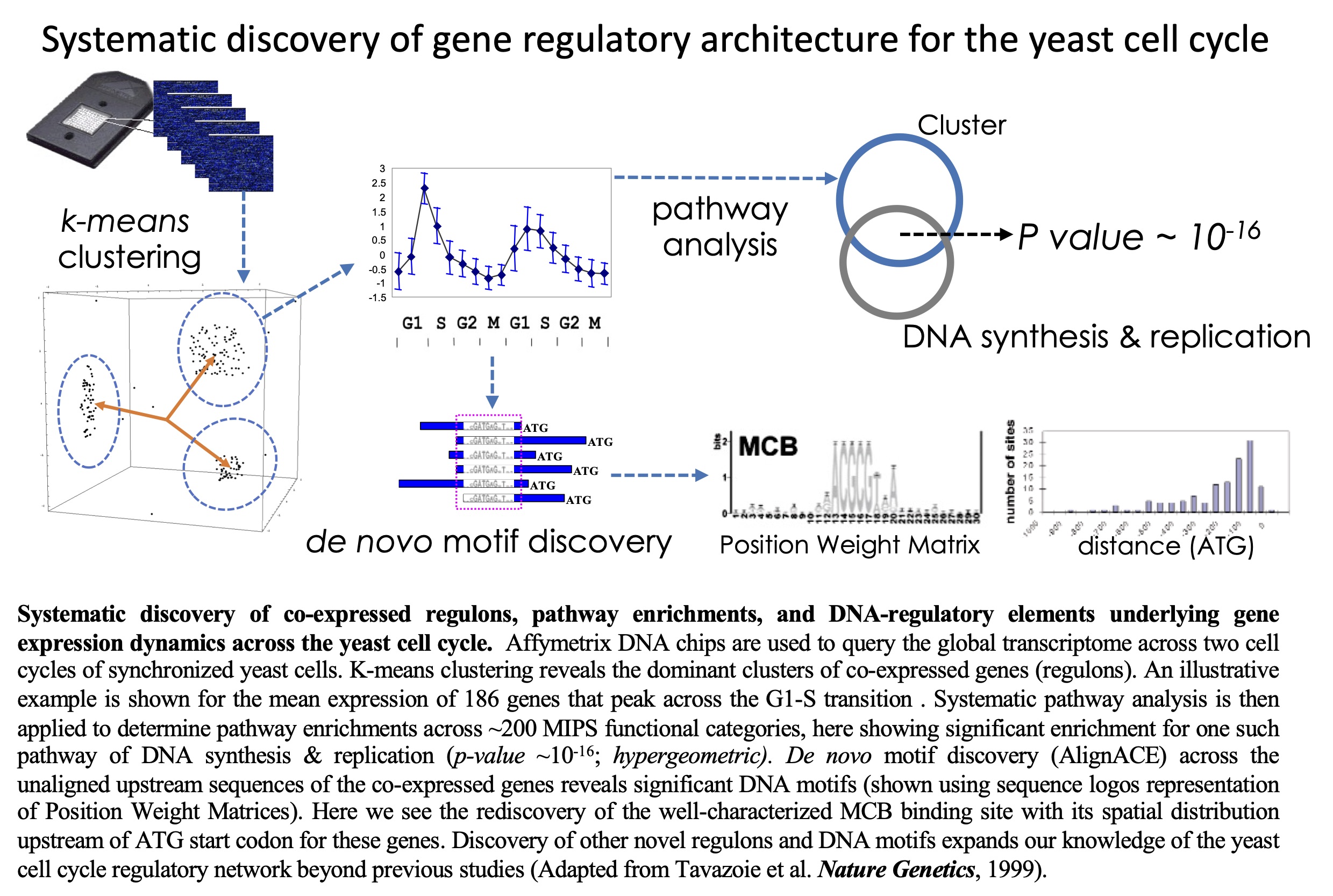
Systematic determination of genetic network architecture.
Nature
Genetics 1999 22:
281-285. PDF
Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM
In
vivo
methylase protection: a technology for global in vivo
monitoring of DNA-protein interactions in bacteria. In this work, we
describe a new technology that enables in vivo monitoring of
DNA-protein interactions in bacteria. Proteins when bound to DNA
block access of DNA methylases to their target sites and this
‘methylase protection’ can be detected using methylation-sensitive
restriction enzymes to reveal in vivo bound sites throughout
the genome. Here, we showed dynamic occupancy patterns of two dozen
sites as a function of genetic and environmental conditions in E.
coli. Our work introduced the notion of clustering analysis on
molecular profiling data, revealing regulons that show common
dynamic patterns across these conditions. Clustering analysis was
subsequently utilized by us and others to discover significant
patterns across a variety of molecular profiling data, including
microarray expression, proteomics, and metabolomics.
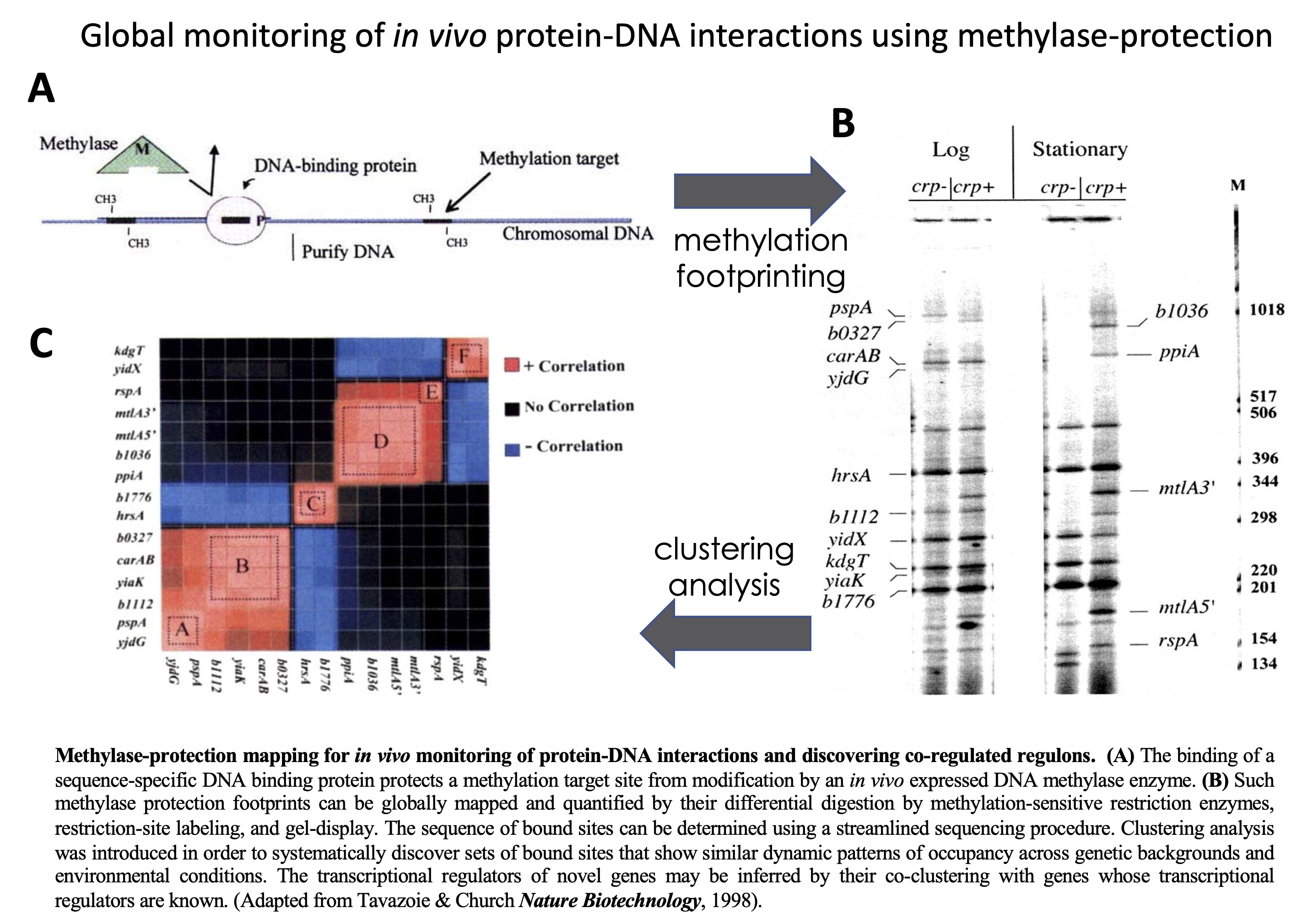
Quantitative whole-genome analysis of DNA-protein interactions by
in vivo methylase protection in E. coli.
Nature
Biotechnology
1998 16: 566-571. PDF
Tavazoie S, Church GM